且不提代价高昂、前途未卜的动物实验及临床试验,仅仅考虑化学部分,还有一个压力巨大的挑战横亘在工艺化学家面前——将药物化学家实验室规模的合成,放大到能支持毒理、药理、药代、分析、制剂研究、临床试验等诸多实验,直到最后的工业化生产规模,同时还要兼顾监管部门的法规要求、知识产权、安全性、环保压力、工艺的稳健性和效率、产品的质量以及最最重要的经济性等方方面面。更麻烦的是,当代药物的选择性和活性越来越高,但分子结构也日趋复杂,这极大的增加了挑战的难度;而且,工艺化学家的时间和资源往往有限。企业上下都期待那些“有前途”的分子结构能早日变成上市挣钱的“明星药物”,工艺化学家的压力可想而知。
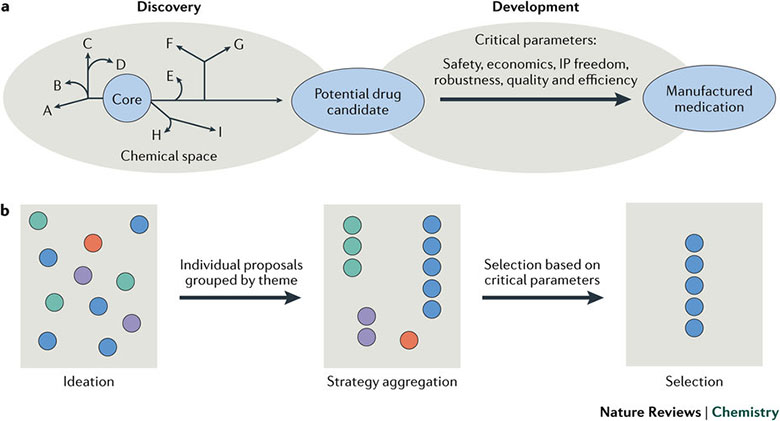
更糟糕的是,因为目的相差很大,不少情况下药物化学家提供的实验室合成路线对工艺化学家的帮助有限,在前者的合成路线上修修补补往往不能让后者达到目标,而是需要进行不破不立的“颠覆性”创新(图1a)。那么,工艺化学家如何进行这种高风险高回报的“颠覆性”创新呢?最近,制药巨头百时美施贵宝(Bristol-Myers Squibb,BMS)的化学和合成研发部门的Martin D. Eastgate等在Nature 的新子刊Nature Reviews Chemistry 上发表综述性文章,结合几个典型案例分析了药企中工艺化学的“颠覆性”创新。除了研发新反应和新工艺,他们还强调了思维扩展-策略聚集-策略筛选的团队工作模式,来减少化学家的个性和偏好对合成路线的影响,以降低“破而后立”过程中的失败风险(图1b)。
图1. a) 化学在药物发现和开发阶段不同的作用,b) 思维扩展-策略聚集-策略筛选的工作模式。
Eastgate建议工艺化学家团队在工作中考虑以下几个问题,以提高成功的几率。
• 我们团队最初如何拆解目标分子?
• 在时间和资源都有限的情况下,我们如何决定哪些合成策略应该优先在实验室中进行尝试?
• 在评估合成建议时,我们如何管理个人偏好?
• 我们如何衡量一个给定策略提高效率的潜力?
• 我们如何确定哪些化学问题需要通过创造性的过程化学方案予以解决,哪些又需要避免?
• 为制定最终商业化的工艺,我们在工艺研发过程中需要哪些概念验证(proof of concepts),哪些又需要放弃?
案例一:先集思广益,后选择合成路线
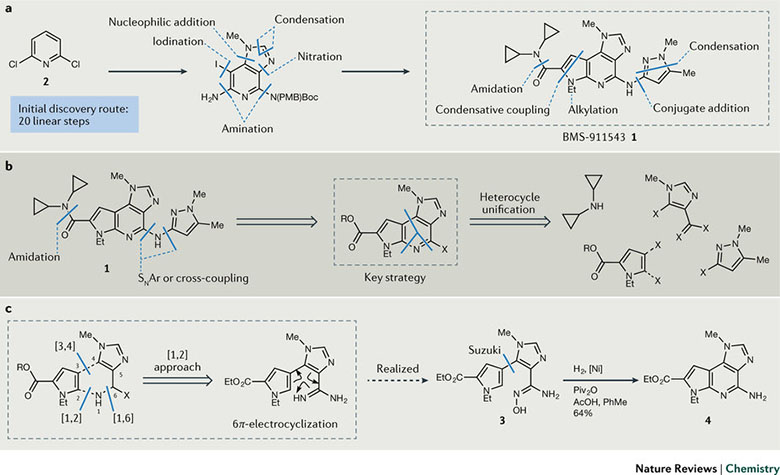
图2. BMS-911543(1)的合成策略。
JAK2抑制剂BMS-911543(1),以2,6-二氯吡啶(2)为起始物料的初始合成路线有20步之多。如此长的路线,对于合成药学相关实验所要求的较大规模的样品肯定不合适,工艺化学家们决定从头设计。与药物发现阶段的以结构多样性为导向不同,工艺化学家的目标更明确——有且只有一个分子。
第一步当然是团队内部“脑风暴”式的思维扩展。尽管多人建议从吡啶衍生物开始,对外围进行官能团化,但这并不会改变路线长而复杂的局面。研发团队开始大破大立,计划将核心的吡啶环拆分为吡咯和咪唑衍生物两个小片段,然后再成环,这看上去符合逻辑,只是形成如此高度官能团化杂环化合物的参考信息相当稀缺,需要花费时间去研究。幸好4个片段的合成已有足够的文献支持,如何将它们连接起来是第一要务,特别是吡咯和咪唑连接后成稠三环母核结构的关键步骤(图2b)。团队可用的时间和资源有限,需要静下心来盘算一下风险和收益比。在三种成环方式中[3,4]-环合反应和[1,2]环合反应首先进行试验,尽管反应机理上看上去可行,但无论是加热还是酸碱催化,均没有成功。团队开创性的采用N自由基、偕胺肟(3)在Ni催化下发生分子内环合反应,然后通过C-H官能化直接生成4(图2c)。关键中间体3可通过3-硼酸酯吡咯和5-溴-4-氰基-1-甲基咪唑通过Suzuki偶联反应制备。最终以7步反应总收率高达22%得到目标产物,相比原来0.05%的总收率提升了440倍,这只用了10个月的时间。
案例二:在专利丛中创新
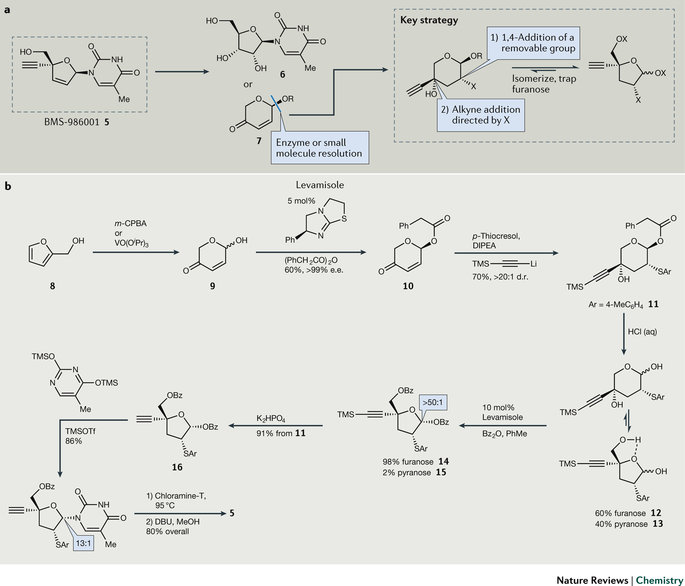
图3. BMS-986001(5)的合成策略。
核苷逆转录酶抑制剂(NRTI)BMS-986001(5)是治疗HIV的候选药物。开发这类药物最大的问题是各种各样的专利,而一个候选药物要想最终商业化,专利保护是个不得不迈过的坎。为合成BMS-986001,工艺化学家团队研究了已有的专利,然后通过逆合成分析提出了两个创新的拆解策略。第一个策略保守些,从容易获得的化合物5-甲基尿苷(6)开始;第二个策略更开放和冒险,从光学纯的吡喃酮(7)出发(图3a)。化学家对两条路线进行了平行的研发,均实现了概念验证,不过后来发现第二条路线更具规模放大的潜力而被重点开发。
最开始的设计中,糠醛(8)经间氯过氧苯甲酸发生Achmatowic氧化,但因废弃物和安全的问题,这一步改用钒类催化剂实现,得到消旋体吡喃酮9。接下来的挑战是手性拆分,借鉴相关类似物的酶拆分,筛选了100多种水解酶,不幸的是绝大多数酶得到的是不想要的R 型产物。最优的酶催化苯甲酸酯水解反应,动力学拆分的选择性高达99.5%,可总收率仅有26%,这时显然要更换方法了。化学家们随即改用亲核催化剂进行动力学拆分,最终筛选了廉价和容易获得的驱虫药左旋咪唑(levamisole)作为催化剂进行动力学不对称酰化反应,化合物9的转化率大于95%,粗品的e.e.为79%。用苯乙酸酐做酰化试剂,产物在重结晶后e.e.达到99%,分离后的产品收率为60%,是酶拆分法的两倍多。化合物10选择性地与对甲苯硫酚反应后,再与(三甲基硅基)乙炔锂发生非对映选择性加成,以70%的收率和优良的非对映选择性(d.r. >20:1)生成11。盐酸水解脱去苯乙酰基得到吡喃糖型的化合物13(95%),将溶剂更换为非极性的甲苯,略微互变为呋喃糖型的12(60 : 40),此时左旋咪唑再次在动力学不对称酰化中立功,端基差向异构体的选择性>50:1(α:β异头物),Vorbrüggen反应插入胸腺嘧啶,在邻位基团芳香基硫醚的辅助下非对映选择性>13:1。接下来,硫醚被氯胺T氧化消除生成双键,甲醇中脱去苄基得到5。从化合物10开始到产品,收率高达44%(图3b)。
在这个案例中,工艺化学家为绕过专利保护,不得不大胆冒险开发新路线,同时开发两条路线。在呋喃酮路线中,他们研究了钒催化呋喃氧化、左旋咪唑的不对称选择性和氯胺消除硫醚等这些不寻常的反应。
案例三:拓展性探索
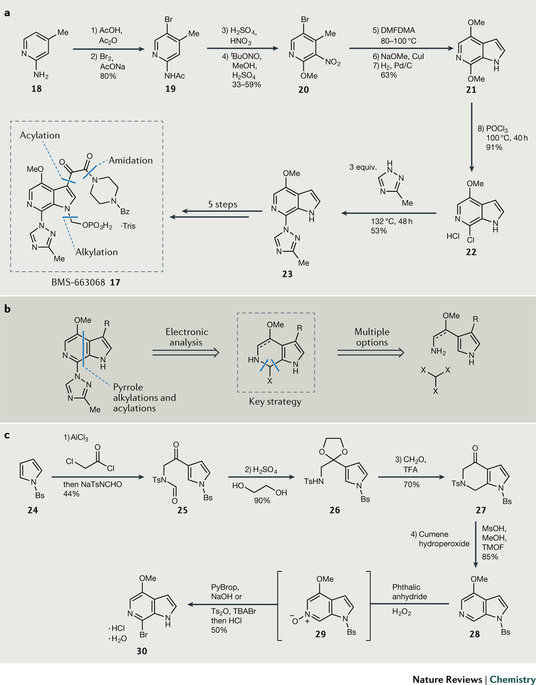
图4. BMS-663068(17)的合成策略。
HIV附着抑制剂BMS-663068(17)进入临床研究,需要大量样品(吨级)。最初的路线以吡啶衍生物18为起始物料,关键中间体为6-氮杂吲哚23。原路线中,纯POCl3的氯化以及芳香族亲核取代反应(SNAr反应)因收率低、大量使用昂贵的三唑等原因不适用于工业化。在这个例子中,因化合物结构新颖,化学家们提出了50多种合成方案,其中大多数还是源于吡啶衍生物。经化学家们冷静分析,这种方式存在很多麻烦,如物料成本高,需要稀有金属催化剂和工艺稳健性不佳。团队决定将氮杂吲哚环的C7和C4之间断裂,重新合成杂环(图4b),这意味着要涉及新的化学研究领域。
以N-苯磺酰基吡咯24为起始物料,C3位经傅克反应乙酰化,但后来发现C3位羰基电子效应的影响导致无法关环,将其修改为缩醛重塑吡咯的亲核性,原位脱去缩醛保护后经Pictet-Spengler环合反应得到27,芳构化为N杂吲哚母核28。现在面临的问题是如何在C7位选择性地官能团化C-H键。氯化产物因后续的SNAr反应差,推测溴化后应该活性更佳,但是当采取类似的氯化反应时,如在POBr3中反应结果不佳。化学家们最终采取了迂回策略,先将28氧化为氮氧化物再溴化,随后铜催化的Ullmann反应偶联三唑。整个反应过程都表现出优秀的选择性和收率,最终10步反应得到产物(图4c)。
整个过程中,思维扩展-策略聚集-策略筛选的团队工作模式起到了决定性的作用。
案例四:挑战不可能
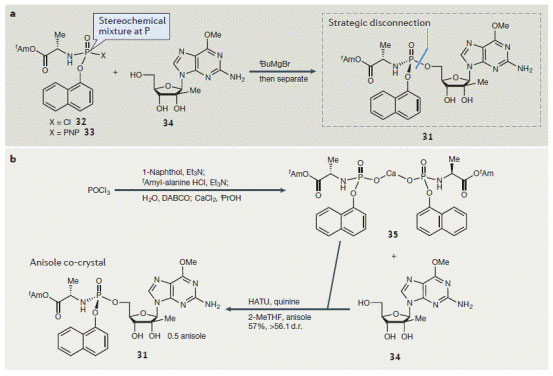
图5. 核苷酸前药(31)的合成策略。
有时候,关键的化学键非常明显,但形成这根键的难度不小,如核苷酸前药31(图5a),最初合成它的反应由Noyori开发,氯亚磷酰胺32和核苷34的烷氧基镁化物反应生成最后的化学键。但活化的磷化合物的物理性质非常折磨人,氯化物32存在1:1的非对映异构体,而且不稳定(在偶联反应条件下迅速降解),这导致31的收率在柱层析后仅有27%。另一种解决方案是采用对硝基苯酚PNP化合物33,它的稳定性好且能结晶,文献报道这类反应中的转化率高。然而由于种种原因这些措施都不理想,不能选择性合成氯或PNP的磷酸酯,还需要额外的设备来拆分。
另一种思路是将磷酰胺酸和核苷34在偶联试剂下成键,如合成多肽时的偶联试剂。团队很快制备出磷酰胺酸,结果大失所望,这是一个粘稠的油状物,实用价值不大。幸好马上出现了转机,自动化筛选发现它的钙盐可以结晶,便于除去杂质和贮藏。在经过高通量筛选偶联试剂、活化试剂和辅助试剂,发现加入手性助剂对于收率和选择性都有积极的影响。优化后,HATU作为偶联试剂,奎宁作为手性助剂,粗品收率提高到89%,选择性为7:1 d.r.,P(S)非对映体为优先产物。该目标异构体在结晶时可高选择性地与苯甲醚形成共晶,得到光学纯的活性药物组分(API),收率达57%。虽然收率属于中等,但这新条路线拥有稳定的中间体、稳健的反应条件,并不需柱层析,相比之前的路线有质的提升。
案例五:物料需求推动研发
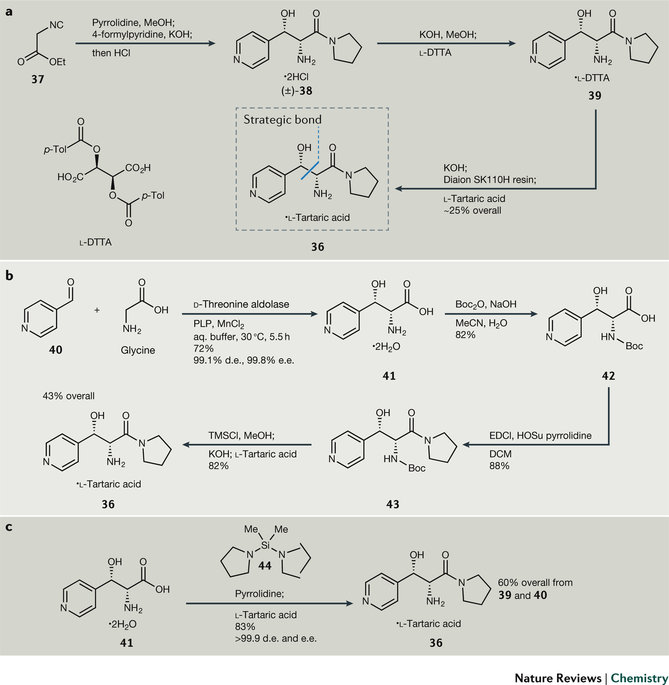
图6. β-羟基氨基酰胺酒石酸盐(36)的合成策略。
β-羟基氨基酰胺酒石酸盐(36)的有效剂量较高(最高约2.7 g/天/人),临床试验的需求量也就很大,达10-12吨。对商业化来说,物料成本和生产周期都是关键指标。因此,工艺化学家积极备战,准备物料,同时开发新的路线以降低成本、提高效率、缩短工期。尽管早期的合成路线也很短(图6a),也存在一些问题不适用于工业化,例如起始物料异腈基乙酸乙酯(37)不稳定、有毒、昂贵、不易购买,以及产物是消旋体,意味着有50%的产品会被浪费。
为解决这个问题,工艺化学家们开发了一种酶催化方法(图6b),实用D-苏氨酸醛缩酶一步反应构建两个手心中心,并且具有优秀的d.e.(99.1%)和e.e.(99.8%)。额外的福利是反应优化后的pH和氨基酸41的等电点接近,因此41直接随反应结晶析出,大大简化了提纯和分离的过程,并且可以使用氨基酸化学的标准程序,例如叔丁基羰基Boc保护基等。这条路线避免使用异腈衍生物,而是用安全廉价的甘氨酸代替,反应总收率也提高至43%。但是,脱保护时出现了麻烦,因为产物也溶于水,无法用常规的水洗操作除去无机盐副产物。另外氨基酰胺36的游离碱对逆向醛醇缩合反应敏感,增加了脱保护基的难度和成本。团队还希望避免引入Boc保护基和脱保护基的方法。为解决这个挑战,团队计划将氨基酸41直接和硅试剂反应,既能保护反应体系也能活化酸,刚开始选用的HMDS未能达成目的,后采用双亲电的硅烷如二氯二甲基硅烷时获得成功。随后,团队另外合成二甲基二吡咯硅烷44进行反应,甲醇淬灭后,蒸出过量的吡咯烷和硅试剂,得到稳定的硅基化产物,加入酒石酸水/醇溶液,硅键断裂,并随着反应进行直接成盐。
在这个案例中,酶催化的使用是个关键,从41一步生成36,减少了废物产生,缩短了时间,并且总收率提高至60%。
案例六:基于机理的化学稳健性
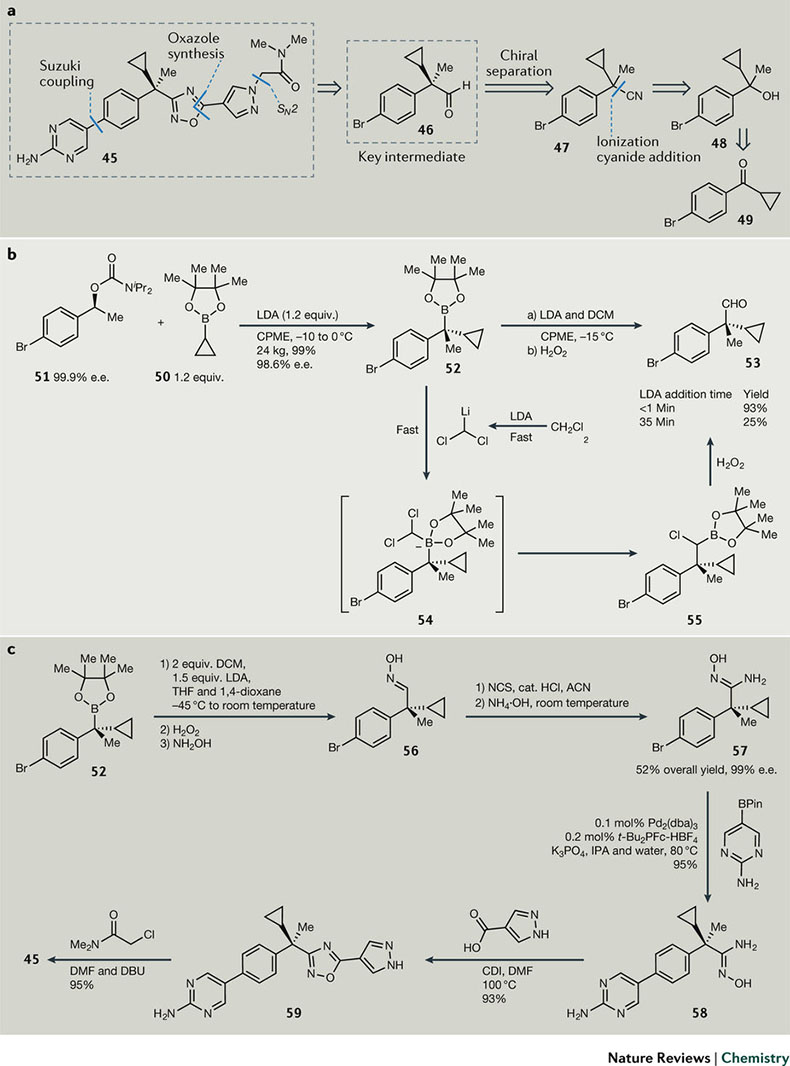
图7. 5-脂氧化酶活化蛋白(FLAP)抑制剂45的合成策略。
有效性和安全性是药物的两大基本要求,这也使当代药物分子的结构越来越复杂,大规模制备的不可预测性和对应参考文献的稀缺,使得工艺化学团队常常要开发新的反应和技术。但工艺路线选择和优化,最好的方式还是基于先前的知识和对反应机理的认识,5-脂氧化酶活化蛋白(FLAP)抑制剂45(图7a)的合成就是这样的案例。45的结构特点是含有一个手性非消旋的季碳立体中心。现在已有多种方式可以构建季碳中心,如Aggrawal的立体选择性硼重排反应。
因为关键的苄基锂化物不稳定,容易失去手性,后发现可能存在底物与二异丙基氨基锂(LDA)的络合物中间态,因此化学家们假设如果锂化物中间体一旦形成就立刻被捕获,可以保留光学纯度,这样即使在工厂中操作时间稍长也不会损失手性。利用LDA与硼酸频哪醇官能团兼容的性质,化学家们进行工艺优化,放大到24 Kg的规模,收率接近定量反应,而且立体化学损失极少(98.6% e.e.,图7b)。经过二氯甲烷脱质子化和同系化反应,得到高光学纯度的中间体53。
现在看来前景一片光明,但问题随后接踵而至,在进行极限条件测试时,发现延长LDA的滴加时间,反应收率急剧下降。滴加时间小于1 min的收率高达93%,而35 min时收率仅有25%。对这条路线而言,LDA的滴加是致命缺陷,并需要基于反应机理的解决方案。对合成硼酸酯54中的LDA拔氢和硼酸酯捕获是快速反应,但55的重排未知。计算发现,反应活化能为17.9 kcal mol-1(B3LYP/6-311G),有可能是限速步骤。现在假设在较高的反应温度下(-15 ℃),中间体55会发生竞争性的重排反应和与DCM阴离子的低聚反应,这样,解决这个问题的方案就变得显而易见了。过渡态中间体54需要保持在合适的温度,直至LDA加完,以屏蔽副反应。又因DCM阴离子极不稳定,容易自己淬灭,反应温度需要增加以促进重排反应,这样温度也不能太低,-25 ℃可以足够满足需求。即使这样,要建立一个可规模化的、经济、安全、稳定的工艺,手性季碳醛53仍是整个路线的拦路虎,需要将其替换。如图7c所示,肟56作为一个固体中间产物,收率52%,且光学纯度达到99% e.e.,再借鉴之前研究的偶联反应、噁二唑杂环成环反应和烷基化反应,终于完成了这个高难度挑战。
案例七:效率驱动的合成设计
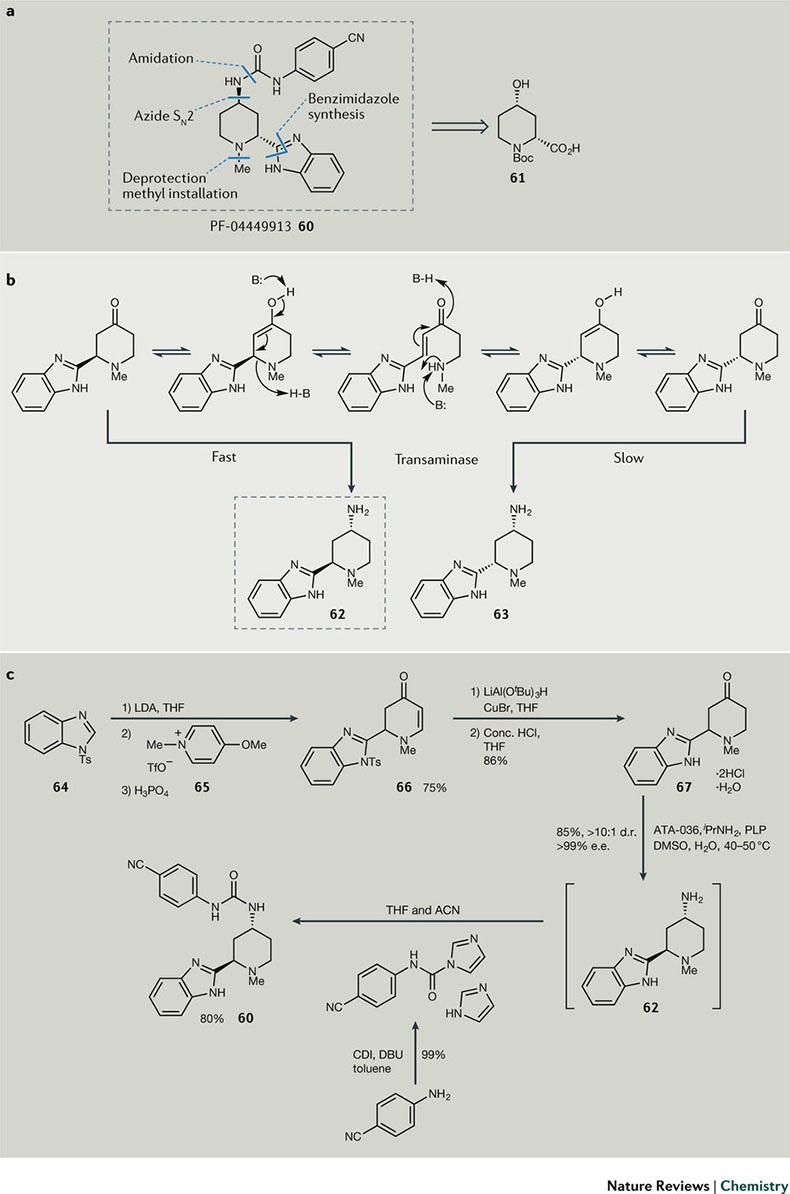
图8. PF-04449913(60)的合成策略。
辉瑞的候选药物PF-04449913(60,图8a)是另一个药物化学家和工艺化学家不同思维方式的案例。Munchhof等通过关键核心化合物4-羟基哌啶酸(61),迅速探索了该结构的化学空间,并发现了高效的抑制剂60,它拥有良好的物理性质。药物发现阶段的合成路线共有14步反应,总收率为4%,且应用了不适合大规模生产的叠氮化学。接力棒交到工艺化学家手中之后,因为药学研发对药物的需求大大增加,因此开发公斤级路线,减少成本势在必行。
生物催化剂,特别是氨基转移酶作为一种高效、安全、绿色且经济的选择,适合大规模合成手性化合物,特别是手性的仲胺化合物。辉瑞的Zhihui Peng和同事们将不对称转胺反应和差向异构2-芳香基-4-哌啶酮结合起来,希望通过动力学拆分同时生成第二个手性中心(图8b)。高风险伴随着高回报,在筛选了多种转胺酶后发现有几种酶可以实现这个目标,以高收率高选择性得到期望的trans 异构体。有这样的关键技术在手,可以顺利进行后续反应。N-对甲苯磺酰基苯并咪唑64在LDA作用下锂化,和活化的吡啶盐65反应得到底物66,共轭加成还原和脱去Ts基团后生成2-苯并咪唑-4-哌啶酮(67),经转氨酶选择性动力学拆分后制备关键中间体62,原位反应后生成药物分子60。这样,合成路线仅为5步,总收率为40%,是原来的10倍,并降低了70%的成本。
这些“颠覆性”创新的案例,无不是化学家们知识、智慧和经验的结晶。这些案例都是在较短的时间内完成使命,解决方案无不充满“颠覆性”的创造性,令人拍案叫绝。无论是否从事工艺化学方向的研究,这些案例都值得我们学习。